Microstructure Quantification
Combining advanced microscopy with AI analytics to quantify drug product microstructures and identify critical quality attributes.


Drug Product Structures
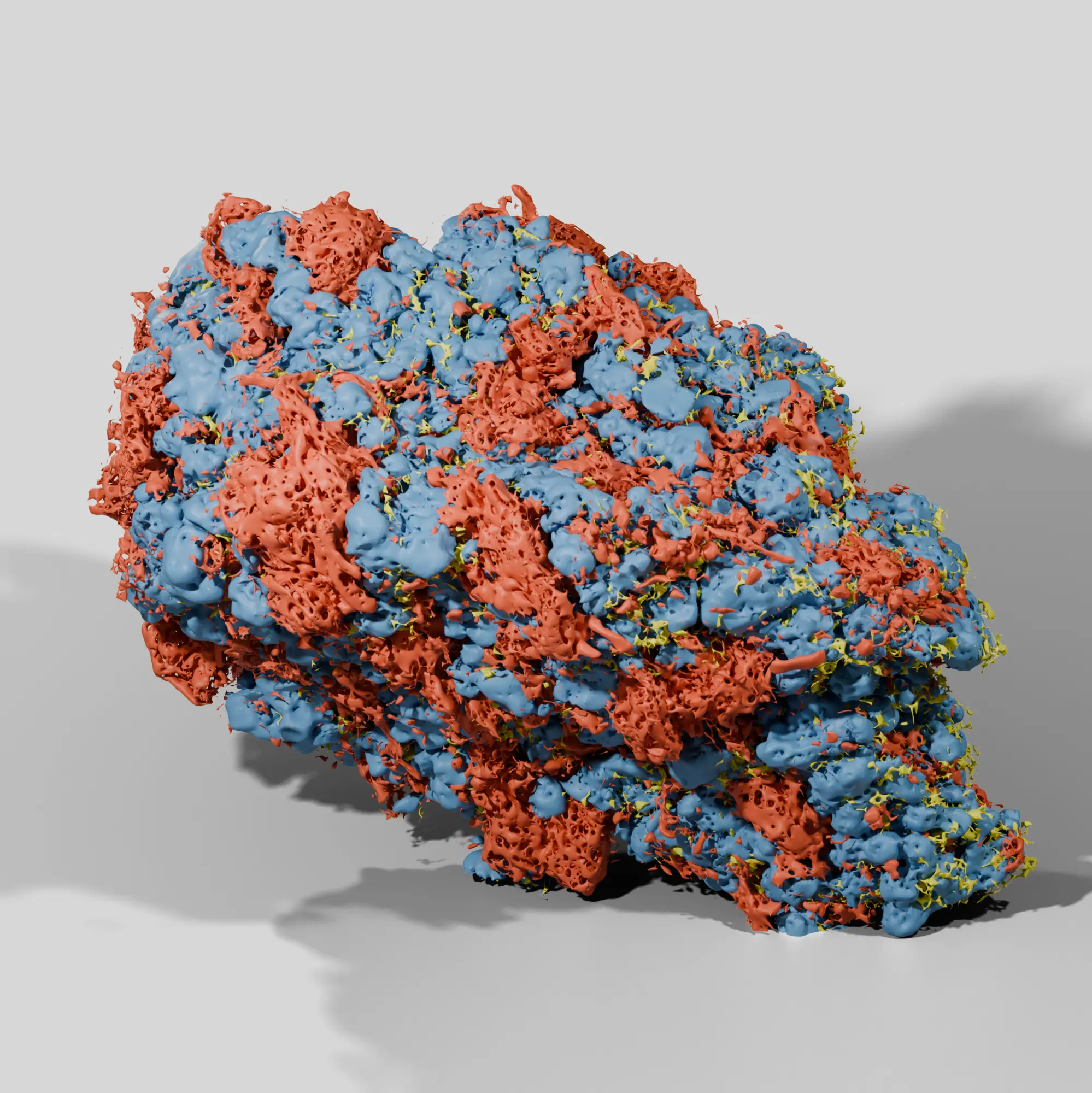
Our process ensures a wholistic analysis of the nanostructures and microstructures behind drug product performance. With advanced microscopy and AI analytics we quantify a library of attributes not readily captured by traditional analytical techniques. Image-based characterization provides a direct and accurate evaluation of microstructures within the final drug product.

Particle Attributes
Particle size is not the only attribute to consider in product development. Other properties like ingredient porosity, surface area, wall thickness, and shape can play a critical role in manufacturability and drug performance. Our all in one analysis provides a full spectrum understanding of these attributes, including properties impossible to measure with routine techniques.

Porosity and Ingredient Compaction
In pharmaceutical development, knowing the porosity amount doesn’t always tell the whole story of product performance and quality. Our image-based workflows reveal what ingredients porosity is associated with, how they change during processing, and their relationship to dissolution and disintegration. We also use digitized pore networks for in silico dissolution.
Transform Your Program with Microstructure Science
Get started with a drug product digital twin.
